
Nội dung chính
Sống khỏe mỗi ngày – Chủ đề: Cơn tăng đường huyết ở bệnh nhân đái tháo đường
Tác giả: ABBAS E. KITABCHI, PHD, MD1 GUILLERMO E. UMPIERREZ, MD2 JOHN M. MILES, MD3 JOSEPH N. FISHER, MD1
Người dịch: BS.Nguyễn Văn Phát
Tải bản đầy đủ Tại đây.
Nhiễm toan ceton do đái tháo đường (DKA) và trạng thái tăng áp lực thẩm thấu do tăng đường huyết(HHS) là hai biến chứng chuyển hóa cấp tính nghiêm trọng nhất của bệnh đái tháo đường. DKA chịu trách nhiệm cho hơn 500.000 ngày nằm viện mỗi năm (1,2) với ước tính chi phí y tế trực tiếp hàng năm và chi phí gián tiếp là 2,4 tỷ USD (2,3). Bảng 1 phác thảo các tiêu chuẩn chẩn đoán DKA và HHS. Bộ ba tăng đường huyết không kiểm soát, nhiễm toan chuyển hóa và tăng tổng nồng độ xeton trong cơ thể đặc trưng cho DKA. HHS được đặc trưng bởi tăng đường huyết nghiêm trọng, tăng áp lực thẩm thấu và mất nước trong trường hợp không có nhiễm toan ceton nghiêm trọng. Những rối loạn chuyển hóa này là kết quả của sự kết hợp của sự thiếu hụt insulin tương đối hoặc tuyệt đối và sự gia tăng các hormone chống điều hòa (glucagon, catecholamine, cortisol và hormone tăng trưởng). Hầu hết bệnh nhân DKA đều mắc bệnh tiểu đường loại 1 tự miễn; tuy nhiên, bệnh nhân tiểu đường loại 2 cũng có nguy cơ trong quá trình stress dị hóa của bệnh cấp tính như chấn thương, phẫu thuật hoặc nhiễm trùng. Tuyên bố đồng thuận này sẽ phác thảo các yếu tố khởi phát và các khuyến cáo để chẩn đoán, điều trị và phòng ngừa DKA và HHS ở đối tượng người lớn. Nó dựa trên một đánh giá khoa học (4) và các bài báo được công bố bởi các chuyên gia gần đây kể từ năm 2001, cần được tham khảo để biết thêm thông tin.
Dịch tễ học
các nghiên cứu dịch tễ học gần đây chỉ ra rằng số ca nhập viện vì DKA ở Hoa Kỳ đang gia tăng. Trong thập kỷ từ 1996 đến 2006, số trường hợp mắc bệnh đã tăng 35%, với tổng số 136.510 trường hợp được chẩn đoán chính là DKA vào năm 2006 — một tốc độ tăng có lẽ nhanh hơn mức tăng tổng thể trong chẩn đoán bệnh tiểu đường (1). Hầu hết bệnh nhân bị DKA ở độ tuổi từ 18 đến 44 (56%) và 45 đến 65 tuổi (24%), chỉ có 18% bệnh nhân <20 tuổi. Hai phần ba bệnh nhân DKA được coi là mắc bệnh tiểu đường loại 1 và 34% mắc bệnh tiểu đường loại 2; 50% là nữ và 45% là da trắng. DKA là nguyên nhân phổ biến nhất gây tử vong ở trẻ em và thanh thiếu niên mắc bệnh tiểu đường loại 1 và chiếm một nửa số ca tử vong ở bệnh nhân đái tháo đường dưới 24 tuổi (5,6). Ở đối tượng người lớn bị DKA, tỷ lệ tử vong chung là <1 % (1); tuy nhiên, tỷ lệ tử vong >5%% đã được báo cáo ở người cao tuổi và ở những bệnh nhân mắc các bệnh đồng thời đe dọa tính mạng (7,8). Tử vong trong những tình trạng này hiếm khi do các biến chứng chuyển hóa của tăng đường huyết hoặc nhiễm toan ceton nhưng liên quan đến bệnh lý khởi phát tiềm ẩn (4,9). Tỷ lệ tử vong do HHS được coi là cao hơn so với tỷ lệ tử vong do DKA, với tỷ lệ tử vong gần đây là 5–20% (10,11). Tiên lượng của cả hai tình trạng này về cơ bản trở nên tồi tệ hơn ở độ tuổi cao nhất với sự hiện diện của hôn mê, hạ huyết áp và các bệnh đi kèm nghiêm trọng (1,4,8, 12,13).
Bệnh học
Các quá trình dẫn đến tăng đường huyết và nhiễm toan ceton được mô tả trong Hình 1 (13). Trong DKA, nồng độ insulin hiệu quả giảm và nồng độ hormone chống điều hòa tăng (catecholamine, cortisol, glucagon và hormone tăng trưởng) dẫn đến tăng đường huyết và nhiễm ceton. Tăng đường huyết phát triển do ba quá trình: tăng tạo gluconeogenesis, tăng tốc độ phân giải glycogen và rối loạn sử dụng glucose của các mô ngoại vi (12–17). Điều này được tăng lên bởi tình trạng kháng insulin thoáng qua do chính sự mất cân bằng hormone cũng như nồng độ axit béo tự do tăng cao (4,18). Sự kết hợp giữa thiếu insulin và tăng các hormone chống điều hoà trong DKA cũng dẫn đến việc giải phóng các axit béo tự do vào hệ tuần hoàn từ mô mỡ (phân giải lipid) và quá trình oxy hóa axit béo không kiểm soát trong gan thành các thể xeton (beta-hydroxybutyrate và acetoace-tate ) (19), với kết quả là xeton máu và nhiễm toan chuyển hóa.
Ngày càng có nhiều bằng chứng chỉ ra rằng tăng đường huyết ở những bệnh nhân bị tăng đường huyết có liên quan đến tình trạng viêm nặng được đặc trưng bởi sự xuất hiện của các cytokine tiền viêm (yếu tố hoại tử khối u alpha và interleu-kin-beta, -6, và -8), Protein phản ứng C, các loại phản ứng oxy hoá và peroxid hóa lipid, cũng như các yếu tố nguy cơ tim
mạch, chất ức chế hoạt hóa plasminogen-1 và các axit béo tự do trong trường hợp không bị nhiễm trùng hoặc bệnh tim mạch (20). Tất cả các thông số này trở về giá trị gần bình thường với liệu pháp insulin và bù dịch trong vòng 24 giờ. Trạng thái tăng đông và tình trạng viêm có thể là do các hiện tượng không đặc hiệu do stress và có thể giải thích một phần mối liên hệ của cơn tăng đường huyết với tình trạng tăng đông máu (21).
Cơ chế bệnh sinh của HHS không được hiểu rõ như DKA, nhưng mức độ mất nước lớn hơn (do bài niệu thẩm thấu) và sự khác biệt về insulin có sẵn để phân biệt với DKA (4,22). Mặc dù sự suy giảm insulin tương đối hiện diện rõ ràng trong HHS, sự tiết insulin nội tiết (phản ánh bởi mức C-peptide) dường như lớn hơn trong DKA, nhưng nó không đáng kể (Bảng 2). Mức độ insulin trong HHS không đủ để tạo điều kiện sử dụng glucose bởi các mô nhạy cảm với insulin nhưng đủ để ngăn ngừa phân giải lipid và tạo ceton sau đó (12).
Các yếu tố khởi phát:
Yếu tố khởi phát phổ biến nhất trong sự phát triển của DKA và HHS là nhiễm trùng (1,4,10). Các yếu tố khởi phát khác bao gồm ngừng hoặc không đủ liệu pháp insulin, viêm tụy, nhồi máu cơ tim, tai biến mạch máu não và thuốc (10,13,14). Ngoài ra, bệnh tiểu đường loại 1 mới khởi phát hoặc ngừng sử dụng insulin ở bệnh tiểu đường loại 1 đã biết trước thường dẫn đến sự phát triển của DKA. Ở những bệnh nhân trẻ mắc bệnh tiểu đường loại 1, các vấn đề tâm lý phức tạp do rối loạn ăn uống có thể là một yếu tố góp phần gây ra 20% trường hợp nhiễm toan ceton tái phát. Các yếu tố có thể dẫn đến thiếu hụt insulin ở bệnh nhân trẻ tuổi bao gồm sợ tăng cân khi kiểm soát trao đổi chất được cải thiện, sợ hạ đường huyết, nổi loạn chống lại người quản lý và stress do bệnh mãn tính.
Trước năm 1993, việc sử dụng các thiết bị truyền insulin dưới da liên tục cũng có liên quan đến tần suất DKA tăng lên (23); tuy nhiên, với sự cải tiến trong công nghệ và giáo dục bệnh nhân tốt hơn, tỷ lệ DKA dường như đã giảm trong những người dùng bơm truyền. Tuy nhiên, các nghiên cứu tiến cứu bổ sung là cần thiết để ghi nhận việc giảm tỷ lệ DKA khi sử dụng các thiết bị truyền insulin dưới da liên tục (24).
Bệnh lý tiềm ẩn kích thích tiết ra các hormone chống điều hòa hoặc làm ảnh hưởng đến đường vào của nước có thể dẫn đến mất nước nghiêm trọng và HHS. Ở hầu hết bệnh nhân HHS, lượng nước uống bị hạn chế là do bệnh nhân nằm liệt giường và trầm trọng hơn do phản ứng khát thay đổi của người cao tuổi. Bởi vì 20% trong số những bệnh nhân này không có tiền sử bệnh tiểu đường, việc nhận biết chậm các triệu chứng tăng đường huyết có thể dẫn đến tình trạng mất nước nghiêm trọng. Người cao tuổi mắc bệnh tiểu đường mới khởi phát (đặc biệt là người bệnh của các cơ sở chăm sóc mãn tính) hoặc những người đã biết mắc bệnh tiểu đường những người trở nên tăng đường huyết và không biết về nó hoặc không thể lấy thêm dịch khi cần thiết sẽ có nguy cơ mắc HHS (10,25).
Các loại thuốc ảnh hưởng đến chuyển hóa carbohydrate, chẳng hạn như corticosteroid, thiazide, chất giống giao cảm và pentamidine, có thể dẫn đến sự phát triển của HHS hoặc DKA (4). Gần đây, một số báo cáo trường hợp chỉ ra rằng thuốc chống loạn thần thông thường cũng như thuốc chống loạn thần không điển hình có thể gây tăng đường huyết và thậm chí DKA hoặc HHS (26,27). Các cơ chế có thể xảy ra bao gồm cảm ứng kháng insulin ngoại vi và ảnh hưởng trực tiếp đến chức năng tế bào tuyến tụy bởi đối kháng thụ thể beta 5-HT1A / 2A / 2C, bằng tác dụng ức chế qua thụ thể alpha2-adrenergic, hoặc do tác dụng gây độc (28).
Ngày càng có nhiều trường hợp DKA mà không có nguyên nhân kết tủa đã xảy ra ở trẻ em, thanh thiếu niên và người lớn mắc bệnh tiểu đường loại 2. Các nghiên cứu quan sát và tiền cứu chỉ ra rằng hơn một nửa số đối tượng người Mỹ gốc Phi và người Mỹ gốc Tây Ban Nha trưởng thành mới được chẩn đoán mắc DKA vô cớ mắc bệnh tiểu đường loại 2 (28-32). Biểu hiện lâm sàng trong những trường hợp này là cấp tính (như trong bệnh tiểu đường loại 1 cổ điển); tuy nhiên, sau một thời gian ngắn điều trị bằng insulin, thường có thể thuyên giảm kéo dài, với việc ngừng điều trị bằng insulin và duy trì kiểm soát đường huyết bằng chế độ ăn
uống hoặc thuốc uống hạ đường huyết. Ở những bệnh nhân như vậy, các đặc điểm lâm sàng và chuyển hóa của bệnh tiểu đường loại 2 bao gồm tỷ lệ béo phì cao, tiền sử gia đình mắc bệnh tiểu đường, dự trữ insulin ở tuyến tụy có thể đo được, tỷ lệ thấp dấu hiệu tự miễn dịch phá hủy tế bào Beta và khả năng ngừng sử dụng liệu pháp insulin trong quá trình theo dõi (28, 31,32). Duy nhất, sự có mặt của yêu cầu insulin thoáng qua sau DKA được ghi nhận chính ở người da đen và người mỹ gốc Tây Ban Nha nhưng cũng được ghi nhận ở người Mỹ, người châu Á và người da trắng. Sự đa dạng của các thể như đái tháo đường type 1 vô căn, đái tháo đường không điển hình, đái tháo đường” Flat búh”, đái tháo đường type 2 thể keton . Một vài nghiên cứu thực nghiệm đã làm sáng tỏ cơ chế bệnh sinh của đái tháo đường type 2 thể ketone . Lúc biểu hiện, họ bị suy giảm rõ rệt sự bài tiết insulin và hoạt động của insulin, nhưng quản lý tích cực với insulin giúp cải thiện hoạt động và bài tiết insulin ở mức tương tự như ở bệnh nhân tiểu đường loại 2 không có DKA (28,31,32). Gần đây, người ta đã báo cáo rằng sự thuyên giảm gần như bình thường hoặc giảm đường huyết có liên quan đến sự phục hồi nhiều hơn của bài tiết insulin cơ bản và kích thích và 10 năm sau khi bệnh tiểu đường khởi phát, 40% bệnh nhân vẫn không phụ thuộc insulin (31). Nồng độ C-peptide lúc đói >1ng / dl (0,33 nmol / l) và mức C-peptide kích thích >1,5ng / dl (0,5 nmol / l) là dự đoán về sự thuyên giảm đường huyết lâu dài ở những bệnh nhân có tiền sử DKA ( 28,32).
Chẩn đoán
Bệnh sử và khám lâm sàng
Quá trình HHS thường tiến triển trong vài ngày đến vài tuần, trong khi diễn biến của đợt DKA cấp tính ở bệnh tiểu đường loại 1 hoặc thậm chí ở bệnh tiểu đường loại 2 có xu hướng ngắn hơn nhiều. Mặc dù các triệu chứng của bệnh tiểu đường được kiểm soát kém có thể xuất hiện trong vài ngày, các thay đổi chuyển hóa điển hình của nhiễm toan ceton thường tiến triển trong một khung thời gian ngắn (thường là <24 giờ). Đôi khi, toàn bộ biểu hiện triệu chứng có thể tiến triển hoặc phát triển cấp tính hơn, và bệnh nhân có thể biểu hiện DKA mà không có manh mối hoặc triệu chứng nào trước đó. Đối với cả DKA và HHS, bệnh cảnh lâm sàng cổ điển bao gồm đa niệu, khát nhiều, sụt cân, nôn mửa, mất nước, suy nhược và thay đổi tình trạng tri giác. Các thăm khám có thể bao gồm nếp véo da mất chậm, nhịp thở Kussmaul (trong DKA), nhịp tim nhanh và hạ huyết áp. Tình trạng tinh thần có thể thay đổi từ tỉnh táo hoàn toàn đến hôn mê hoặc hôn mê sâu, tình trạng nặng hơn thường xuyên gặp trong HHS. Các dấu hiệu thần kinh khu trú (bán manh và liệt nửa người) và co giật (khu trú hoặc toàn thể) cũng có thể là đặc điểm của HHS (4,10). Mặc dù nhiễm trùng là một yếu tố kết tủa phổ biến cho cả DKA và HHS, bệnh nhân có thể nhiệt độ bình thường hoặc thậm chí hạ thân nhiệt chủ yếu do giãn mạch ngoại vi. Hạ thân nhiệt nghiêm trọng, nếu có, là một dấu hiệu tiên lượng xấu (33). Buồn nôn, nôn, đau bụng lan tỏa thường gặp ở bệnh nhân DKA (>50%) nhưng không phổ biến ở HHS (33). Cần thận trọng với bệnh nhân những người phàn nàn về đau bụng khi xuất hiện vì các triệu chứng có thể là kết quả của DKA hoặc dấu hiệu của nguyên nhân khởi phát của DKA, đặc biệt ở những bệnh nhân trẻ hơn hoặc trong trường hợp không bị nhiễm toan chuyển hóa nặng (34,35). Đánh giá thêm là cần thiết nếu phàn nàn này không giải quyết với sự giải quyết tình trạng mất nước và nhiễm toan chuyển hóa.
Cận lâm sàng
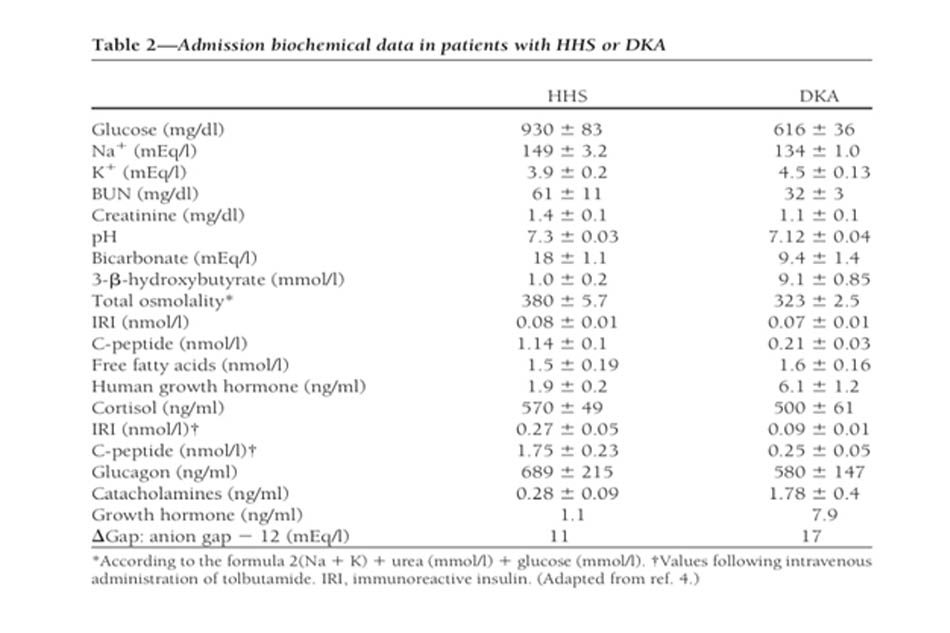
Tiêu chuẩn chẩn đoán DKA và HHS được thể hiện trong Bảng 1. Đánh giá ban đầu của bệnh nhân bao gồm xác định glucose huyết tương, nitơ urê máu, creatinin, chất điện giải ( tính toán khoảng trống anion ),áp lực thẩm thấu máu, ceton huyết thanh và nước tiểu, và phân tích nước tiểu, như cũng như khí máu động mạch ban đầu và công thức máu hoàn chỉnh với sự khác biệt. Cũng nên lấy điện tâm đồ, chụp X-quang phổi và cấy nước tiểu, đờm hoặc máu.
Mức độ nghiêm trọng của DKA được phân loại là nhẹ, trung bình hoặc nặng dựa trên mức độ nghiêm trọng của nhiễm toan chuyển hóa (pH máu, bicarbonate và xeton) và sự hiện diện của tình trạng tâm thần thay đổi (4). Sự chồng chéo đáng kể giữa DKA và HHS đã được báo cáo ở hơn một phần ba số bệnh nhân (36). Mặc dù hầu hết bệnh nhân HHS có pH nhập viện >7,30 và mức bicarbonat >18mEq / l, có thể có ceton huyết nhẹ (4,10).
Tăng đường huyết nghiêm trọng và mất nước với tình trạng tâm thần thay đổi khi không có nhiễm toan đáng kể là đặc điểm của HHS, biểu hiện lâm sàng với ít nhiễm ceton hơn và tăng đường huyết nhiều hơn DKA. Điều này có thể là do nồng độ insulin trong huyết tương (như được xác định bởi C-peptide cơ bản và được kích thích [Bảng 2]) đủ để ngăn ngừa quá trình phân giải lipid quá mức và tạo ceton sau đó nhưng không làm tăng đường huyết (4).
Đặc điểm chẩn đoán chính trong DKA là sự gia tăng nồng độ xeton tổng trong máu lưu hành. Đánh giá tăng ceton máu thường được thực hiện bằng phản ứng nitroprusside, phản ứng ước tính bán định lượng mức acetoacetate và aceton. Mặc dù xét nghiệm nitroprusside (cả trong nước tiểu và huyết thanh) có độ nhạy cao, nó có thể đánh giá thấp mức độ nghiêm trọng của nhiễm toan ceton vì xét nghiệm này không nhận ra sự hiện diện của beta-hydroxybutyrat, sản phẩm chuyển hóa chính trong nhiễm toan ceton (4,12). Nếu có, phép đo beta- hydroxybutyrate huyết thanh có thể hữu ích cho chẩn đoán (37). Sự tích tụ ceton dẫn đến nhiễm toan chuyển hoá tăng khoảng trống anion. Khoảng trống anion được tính bằng cách lấy tổng nồng độ natri trừ đi tổng nồng độ clorua và bicacbonat: [Na – (Cl + HCO3)]. Khoảng trống anion bình thường là từ 7 đến 9 mEq / l và khoảng trống anion >10–12 mEq / l cho thấy sự hiện diện của tăng toan chuyển hóa khoảng trống anion (4).
Tăng đường huyết là một tiêu chuẩn chẩn đoán chính của DKA; tuy nhiên, có thể có một lượng lớn glucose huyết tương khi nhập viện. Các nghiên cứu ngắn gọn về tốc độ sản xuất glucose ở gan đã báo cáo tốc độ dao động từ bình thường hoặc gần bình thường (38) đến cao (12,15), có thể góp phần vào phạm vi rộng của mức đường huyết trong DKA mà không phụ thuộc vào mức độ nghiêm trọng của nhiễm toan ceton (37) . Khoảng 10% dân số DKA có cái gọi là “DKA euglycemic” – mức đường huyết <250mg / dl (38). Điều này có thể là do sự kết hợp của các yếu tố, bao gồm tiêm insulin ngoại sinh trên đường đến bệnh viện, hạn chế thức ăn trước (39, 40) và ức chế tạo gluconeogenesis.
Khi nhập viện, tăng bạch cầu với số lượng tế bào trong khoảng 10.000 –15.000 mm3 thường trong DKA và có thể không phải là dấu hiệu của nhiễm trùng.. Tuy nhiên, tăng bạch cầu với số lượng tế bào>25.000 mm3 có thể chỉ định nhiễm trùng và cần đánh giá thêm (41). Trong nhiễm toan ceton, tăng bạch cầu được cho là do căng thẳng và có thể liên quan đến nồng độ cortisol và norepinephrine tăng cao (42). Natri huyết thanh nhập viện thường thấp do dòng chảy theo chiều thẩm thấu của nước từ nội bào ra ngoài tế bào khi có tăng đường huyết. Nồng độ natri huyết thanh tăng hoặc thậm chí bình thường khi có tăng đường huyết cho thấy mức độ mất nước tự do khá lớn. Để đánh giá mức độ nghiêm trọng của thiếu hụt natri và nước, natri huyết thanh có thể được điều chỉnh bằng cách thêm 1,6 mg / dl vào natri huyết thanh đo được cho mỗi 100 mg / dl glucose trên 100 mg / dl (4,12).
Các nghiên cứu về độ thẩm thấu huyết thanh và sự thay đổi tinh thần đã thiết lập một mối quan hệ tuyến tính thuận giữa nồng độ thẩm thấu và sự suy giảm tri giác (9,36). Sự xuất hiện của tình trạng choáng váng hoặc hôn mê ở bệnh nhân đái tháo đường khi không có nồng độ thẩm thấu hiệu quả (>=320mOsm / kg) đòi hỏi phải xem xét ngay các nguyên nhân khác của sự thay đổi trạng thái tri giác. Trong tính toán độ thẩm thấu hiệu quả, [ion natri (mEq / l) x 2 + glucose (mg / dl) / 18], nồng độ urê không được tính đến vì nó có thể thẩm thấu tự do và sự tích tụ của nó không gây ra những thay đổi lớn về thể tích nội bào hoặc gradient thẩm thấu qua màng tế bào (4).
Nồng độ kali huyết thanh có thể tăng do sự chuyển dịch kali ra bên ngoài do thiếu insulin, ưu trương và tăng hoá máu (43). Bệnh nhân có nồng độ kali huyết thanh bình thường hoặc thấp khi nhập viện có tình trạng thiếu kali toàn cơ thể trầm trọng và cần theo dõi cẩn thận và bù kali mạnh hơn vì điều trị làm giảm kali hơn nữa và có thể gây rối loạn nhịp tim. “Giả đường huyết bình thường “(44) và” giả hạ natri máu “ (45) có thể xảy ra trong DKA khi có nhũ trấp máu nặng.
Nồng độ phosphat huyết thanh nhập viện ở bệnh nhân DKA, như kali huyết thanh, thường tăng cao và không phản ánh tình trạng thiếu hụt thực sự của cơ thể do sự dịch chuyển đồng nhất của phosphat nội bào sang ngoại bào (12, 46,47). Sự thiếu hụt insulin, ưu trương và tăng dị hóa đều góp phần vào sự di chuyển của phosphate ra khỏi tế bào.
Tăng amylase máu đã được báo cáo ở 21–79% bệnh nhân bị DKA (48); tuy nhiên, có rất ít mối tương quan giữa sự hiện diện, mức độ, hoặc loại isoenzyme của tăng amylase máu và sự hiện diện của các triệu chứng tiêu hóa (buồn nôn, nôn và đau bụng) hoặc các nghiên cứu hình ảnh tuyến tụy (48). Định lượng lipase huyết thanh có thể có lợi trong chẩn đoán phân biệt với viêm tụy; tuy nhiên, lipase cũng có thể tăng trong DKA nếu không có viêm tụy (48).
Chẩn đoán phân biệt
Không phải tất cả bệnh nhân nhiễm toan ceton đều có DKA. Nhiễm ceton do đói và nhiễm toan ceton do rượu được phân biệt theo tiền sử lâm sàng và nồng độ glucose trong huyết tương từ tăng nhẹ (hiếm khi >200mg / dl) đến hạ đường huyết (49). Ngoài ra, mặc dù nhiễm toan ceton do rượu có thể dẫn đến nhiễm toan nặng, nhưng nồng độ bicarbonate huyết thanh trong nhiễm ceton do đói thường không phải là <18mEq / l. DKA cũng phải được phân biệt với các nguyên nhân khác của nhiễm toan chuyển hóa có khoảng trống anion cao, bao gồm cả nhiễm toan lactic; ngộ độc như salicylate, methanol, ethylene glycol, và paraldehyde; và đợt cấp suy thận mạn (4). Vì tình trạng nhiễm axit lactic phổ biến ở bệnh nhân đái tháo đường hơn ở người không bị đái tháo đường và vì nồng độ axit lactic tăng cao có thể xảy ra ở những bệnh nhân thiếu hụt thể tích nghiêm trọng, nên đo lactate máu khi nhập viện.
Cần tìm tiền sử lâm sàng của việc lạm dụng thuốc trước đây. Đo nồng độ salicylat trong huyết thanh và nồng độ metanol trong máu có thể hữu ích. Ethylene glycol (chống đóng băng) được gợi ý bởi sự hiện diện của các tinh thể canxi oxalat và hipprat trong nước tiểu. Việc nuốt phải paraldehyde được biểu hiện bằng mùi đặc trưng của nó trong hơi thở. Bởi vì những chất gây độc này là các hợp chất hữu cơ có trọng lượng phân tử thấp, chúng có thể tạo ra khoảng trống thẩm thấu ngoài nhiễm axit khoảng trống anion (14). Một báo cáo gần đây nói rằng việc sử dụng cocaine là một yếu tố nguy cơ độc lập đối với DKA tái phát (50).
Gần đây, một báo cáo trường hợp đã chỉ ra rằng một bệnh nhân mắc bệnh to cực được chẩn đoán có thể có DKA là biểu hiện chính của bệnh (51). Ngoài ra, một báo cáo trước đó về chứng to tuyến yên đã được trình bày với hai đợt DKA với sự phục hồi hoàn toàn bệnh đái tháo đường sau xuất huyết tuyến yên (52).
Điều trị
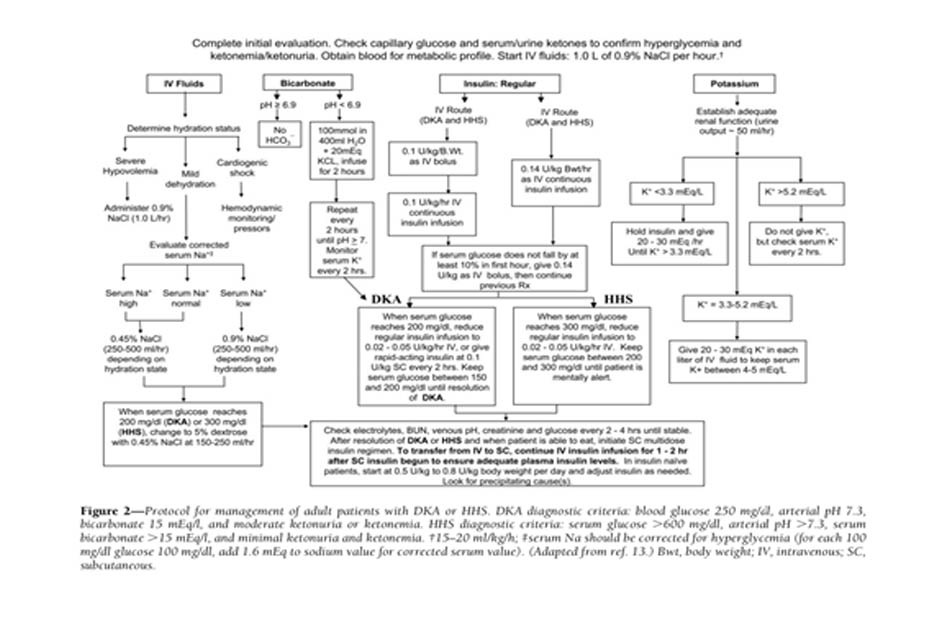
Điều trị thành công DKA và HHS đòi hỏi phải điều chỉnh tình trạng mất nước, tăng đường huyết và mất cân bằng điện giải; xác định yếu tố khởi phát đồng mắc; và trên hết là theo dõi bệnh nhân thường xuyên. Các phác đồ quản lý bệnh nhân DKA và HHS được tóm tắt trong Hình 2 (52).
Bù dịch
Bù dịch ban đầu hướng đến việc mở rộng thể tích nội mạch, mô kẽ và nội bào, tất cả đều được giảm trong các cơn tăng đường huyết (53) , và phục hồi tưới máu thận. Trong trường hợp không có tổn thương tim, nước muối đẳng trương (0,9% NaCl) được truyền với tốc độ 15-20ml/kg/h, 1–1.5l trong giờ đầu tiên. Lựa chọn bù dịch tiếp theo phụ thuộc vào huyết động học, tình trạng mất nước, nồng độ điện giải và lượng nước tiểu. Nói chung, truyền NaCl 0,45% ở 250 –500 ml / h là thích hợp nếu natri huyết thanh hiệu chỉnh là bình thường hoặc tăng cao; NaCl 0,9% với tốc độ tương tự là thích hợp nếu natri huyết thanh hiệu chỉnh thấp (Hình 2). Sự tiến triển thành công của việc bù dịch được đánh giá bằng theo dõi huyết động (cải thiện huyết áp), đo lượng dịch đầu vào / đầu ra, giá trị xét nghiệm và khám lâm sàng.
Việc bù dịch để điều chỉnh các khoản thiếu hụt ước tính trong vòng 24 giờ đầu tiên. Ở những bệnh nhân bị tổn thương thận hoặc tim, phải theo dõi nồng độ thẩm thấu của huyết thanh và thường xuyên đánh giá tình trạng tim, thận và tri giác trong quá trình hồi sức truyền dịch để tránh quá tải dịch (4,10, 15,53). Sự bù dịch tích cực và sau đó là điều chỉnh áp lực thẩm tháu máu đã được chứng minh là dẫn đến đáp ứng mạnh mẽ hơn với liệu pháp insulin liều thấp (54).
Trong khi điều trị DKA, tình trạng tăng đường huyết được điều chỉnh nhanh hơn so với ketoacido. Thời gian điều trị trung bình cho đến khi đường huyết là 250 mg / dl và tình trạng nhiễm ceton (pH>7,30; bicarbonat >18mmol / l) được điều chỉnh tương ứng là 6 và 12 giờ (9,55). Khi glucose huyết tương là <200 mg / dl, nên thêm dextrose 5% vào dịch thay thế để cho phép tiếp tục sử dụng insulin cho đến khi kiểm soát được ceton máu đồng thời tránh hạ đường huyết.
Insulin
Phương pháp chính trong điều trị DKA liên quan đến việc sử dụng insulin regular qua truyền tĩnh mạch liên tục hoặc bằng cách tiêm dưới da hoặc tiêm bắp thường xuyên (4,56,57). Các nghiên cứu ngẫu nhiên có đối chứng ở bệnh nhân DKA đã chỉ ra rằng liệu pháp insulin có hiệu quả bất kể đường dùng thuốc nào (47). Việc truyền insulin regular tĩnh mạch liên tục là cách được ưu tiên vì thời gian bán hủy ngắn và dễ chỉnh liều, và khởi đầu tác dụng chậm và kéo dài thời gian bán hủy của insulin dưới da (36,47,58).
Nhiều nghiên cứu ngẫu nhiên tiến cứu đã chứng minh rằng việc sử dụng insulin regular liều thấp truyền tĩnh mạch là đủ để phục hồi thành công bệnh nhân DKA. Cho đến gần đây, các thuật toán điều trị khuyến cáo sử dụng liều insulin bolus ban đầu (0,1 đơn vị / kg) sau đó truyền 0,1 đơn vị / kg /h (Hình 2). Một nghiên cứu ngẫu nhiên tiếu cứu gần đây đã báo cáo rằng Liều bolus insulin không cần thiết nếu bệnh nhân được truyền insulin 0,14 đơn vị / kg thể trọng / giờ(tương đương 10 đơn vị / giờ ở bệnh nhân 70 kg) (59). Tuy nhiên, trong trường hợp không có liều bolus ban đầu, liều 0,1UI/kg/h dẫn đến nồng độ insulin thấp hơn, nồng độ này có thể không đủ để ức chế cơ thể sản xuất xeton ở gan mà không có liều bổ sung insulin (15).
Các phác đồ truyền insulin liều thấp làm giảm nồng độ glucose huyết tương với tốc độ 50–75 mg/dl /h. Nếu glucose huyết tương không giảm 50–75 mg/dl so với giá trị ban đầu trong giờ đầu tiên, nên tăng liều insulin mỗi giờ cho đến khi đạt được mức giảm glucose ổn định (Hình 2). Khi glucose huyết tương đạt 200 mg / dl trong DKA hoặc 300 mg / dl trong HHS, có thể giảm tốc độ truyền insulin xuống 0,02– 0,05 đơn vị/kg/h, lúc đó dextrose có thể được thêm vào dịch truyền tĩnh mạch (Hình 2). Sau đó, tốc độ sử dụng insulin hoặc nồng độ dextrose có thể cần được điều chỉnh để duy trì các giá trị glucose trong khoảng 150 đến 200 mg / dl trong DKA hoặc 250 và 300 mg / dl trong HHS cho đến khi chúng được hồi phục.
Điều trị bằng các insulin tác dụng nhanh tiêm dưới da (lispro và aspart) đã được chứng minh là một biện pháp thay thế hiệu quả cho việc sử dụng truyền insulin tĩnh mạch trong điều trị DKA. Điều trị bệnh nhân bị DKA mức độ nhẹ và trung bình bằng các insulin tác dụng nhanh tiêm dưới da cứ sau 1 hoặc 2 giờ tại các cơ sở không phải chăm sóc đặc biệt đã được chứng minh là an toàn và hiệu quả như điều trị bằng insulin truyền tĩnh mạch trong ICU ( 60,61). Tốc độ giảm nồng độ glucose trong máu và thời gian điều trị trung bình cho đến khi điều chỉnh được tình trạng nhiễm toan là tương tự nhau ở những bệnh nhân được điều trị bằng các insulin tiêm dưới da cứ sau 1 hoặc 2 giờ hoặc với insulin truyền tĩnh mạch. Tuy nhiên, cho đến khi những nghiên cứu này được xác nhận bên ngoài phạm vi nghiên cứu, những bệnh nhân bị DKA nặng, hạ heuyết áp, phù toàn thân, hoặc bệnh nền nặng nên được quản lý bằng insulin truyền tĩnh mạch trong ICU.
Kali
Bất chấp tình trạng thiếu huỵ kali toàn cơ thể, tăng kali máu mức độ nhẹ đến trung bình thường gặp ở những bệnh nhân bị tăng đường huyết. Liệu pháp insulin, điều chỉnh tình trạng nhiễm toan và tăng thể tích làm giảm nồng độ kali huyết thanh. Để ngăn ngừa hạ kali máu, việc bù kali được bắt đầu sau khi nồng độ trong huyết thanh giảm xuống dưới mức giới hạn trên của giá trị bình thường cụ thể (5,0 –5,2 mEq / l). Mục tiêu điều trị là duy trì nồng độ kali huyết thanh trong giới hạn bình thường 4 –5 mEq / l. Nói chung, 20–30 mEq kali trong mỗi lít dịch truyền là đủ để duy trì nồng độ kali huyết thanh trong giới hạn bình thường. Hiếm khi bệnh nhân DKA có thể bị hạ kali máu đáng kể. Trong những trường hợp như vậy, việc bù kali nên bắt đầu bằng liệu pháp truyền dịch, và điều trị insulin nên được trì hoãn cho đến khi nồng độ kali được khôi phục về>3,3 mEq / l để tránh loạn nhịp tim đe dọa tính mạng và yếu cơ hô hấp (4,13).
Bicarbonate
Việc sử dụng bicarbonate trong DKA còn gây tranh cãi (62) vì hầu hết các chuyên gia tin rằng trong quá trình điều trị, khi lượng ceton giảm xuống sẽ có đủ bicarbonat trừ những bệnh nhân nhiễm toan nặng. Nhiễm toan chuyển hóa nghiêm trọng có thể dẫn đến suy giảm co bóp cơ tim, giãn mạch não và hôn mê, và một số biến chứng đường tiêu hóa (63). Một nghiên cứu ngẫu nhiên tiến cứu ở 21 bệnh nhân không cho thấy những thay đổi có lợi hoặc có hại về tỷ lệ mắc bệnh hoặc tử vong khi điều trị bằng bicarbonate ở những bệnh nhân DKA có pH động mạch nhập viện từ 6,9 đến 7,1 (64). Chín nghiên cứu nhỏ trên tổng số 434 bệnh nhân nhiễm toan ceton do đái tháo đường (217 bệnh nhân được điều trị bằng bicacbonate và 178 bệnh nhân không điều trị bằng kiềm [62]) ủng hộ quan điểm rằng liệu pháp bicacbonate cho DKA không mang lại lợi ích gì trong việc cải thiện tim hoặc các chức năng thần kinh hoặc tốc độ phục hồi của tình trạng tăng đường huyết và tăng ceton huyết. Hơn nữa, một số tác dụng có hại của liệu pháp bicarbonate đã được ghi nhận, chẳng hạn như tăng nguy cơ hạ đường huyết, giảm hấp thu oxy ở mô (65), phù não (65) và phát triển nhiễm toan hệ thần kinh trung ương đảo ngược
Không có nghiên cứu ngẫu nhiên tiến cứu nào liên quan đến việc sử dụng bicarbonate trong DKA với giá trị pH <6,9 đã được báo cáo (66). Vì tình trạng nhiễm toan nặng có thể dẫn đến nhiều tác dụng phụ đối với mạch máu (63), khuyến cáo rằng bệnh nhân người lớn có độ pH <6,9 nên dùng 100 mmol natri bicarbonat (hai ống) trong 400 ml nước vô trùng (dung dịch đẳng trương) với 20 mEq KCI được truyền với tốc độ 200 ml / giờ trong 2 giờ cho đến khi pH tĩnh mạch là >7,0. Nếu độ pH vẫn còn <7,0 sau khi truyền, chúng tôi khuyên bạn nên truyền lặp lại sau mỗi 2 giờ cho đến khi pH đạt đến >7,0 (Hình 2)
Phosphate
Mặc dù thiếu hụt phosphat toàn cơ thể trong DKA trung bình 1,0 mmol / kg trọng lượng cơ thể, nhưng phosphat huyết thanh thường biểu hiện bình thường hoặc tăng . Nồng độ phosphat giảm khi điều trị bằng insulin. Các nghiên cứu ngẫu nhiên tiến cứu đã không cho thấy bất kỳ tác dụng có lợi nào của việc bù phosphat đối với kết quả lâm sàng ở DKA (46,67), và điều trị quá liều phosphat có thể gây hạ calci huyết nghiêm trọng (46,68). Tuy nhiên, để tránh suy cơ tim và cơ xương và ức chế hô hấp do giảm phosphat máu, đôi khi có thể chỉ định thay thế phosphat cẩn thận ở những bệnh nhân bị rối loạn chức năng tim, thiếu máu hoặc suy hô hấp và ở những người có nồng độ phosphat huyết thanh<1,0 mg / dl (4, 12). Khi cần, có thể bổ sung 20 –30 mEq / l kali photphat vào dịch thay thế. Tốc độ thay thế phosphat tối đa thường được coi là an toàn để điều trị giảm phosphat máu nặng là 4,5 mmol / h (1,5 ml / h K2 PO4) (69). Không có nghiên cứu nào về việc sử dụng phosphate trong điều trị HHS.
Chuyển sang insulin dưới da
Bệnh nhân DKA và HHS nên được điều trị bằng insulin tiêm tĩnh mạch liên tục cho đến khi tình trạng tăng đường huyết được giải quyết. Tiêu chuẩn để giải quyết tình trạng nhiễm toan ceton bao gồm đường huyết <200mg / dl và hai trong số các tiêu chuẩn sau: mức bicarbonat huyết thanh >=15mEq / l, pH tĩnh mạch >7.3, và khoảng trống anion tính được <=12mEq / l.Sự giải quyết của HHS có liên quan đến áp lực thẩm thấu bình thường và phục hồi trạng thái tri giác bình thường. Khi điều này xảy ra, liệu pháp insulin dưới da có thể được bắt đầu. Để ngăn ngừa tái phát tăng đường huyết hoặc nhiễm toan ceton trong giai đoạn chuyển tiếp sang insulin tiêm dưới da, điều quan trọng là phải tiếp tục 1-2h insulin truyền tĩnh mạch 1-2h sau khi bắt đầu insulin dưới da. Nếu bệnh nhân vẫn không ăn được, tốt hơn là nên tiếp tục truyền insulin tĩnh mạch và bù dịch. Bệnh nhân mắc bệnh tiểu đường đã biết có thể được cung cấp insulin với liều lượng họ đã dùng trước khi bắt đầu DKA miễn là nó kiểm soát được lượng đường thích hợp. Ở những bệnh nhân chưa từng sử dụng insulin, nên bắt đầu chế độ đa liều insulin với liều 0,5–0,8 đơn vị /kg/ ngày (13). Insulin người (NPH và regular) thường được tiêm hai hoặc ba liều mỗi ngày. Gần đây hơn, các phác đồ nền -bolus với nền (glargine và detemir) và insulin tác dụng nhanh (lispro, aspart, hoặc glulisine) đã được đề xuất như một chế độ insulin sinh lý hơn ở bệnh nhân có đường loại 1. Một thử nghiệm ngẫu nhiên tiến cứu so sánh việc điều trị với chế độ nền -bolus, bao gồm glargine một lần mỗi ngày và glulisine trước bữa ăn, với một chế độ hỗn hợp chia nhỏ của NPH cộng với insulin regular hai lần mỗi ngày sau khi giải quyết DKA. Sự chuyển đổi sang glargine và glulisine dưới da dẫn đến kiểm soát đường huyết tương tự so với NPH và insulin regular; tuy nhiên, điều trị bằng bolus cơ bản có liên quan đến tỷ lệ biến cố hạ đường huyết thấp hơn (15%) so với tỷ lệ ở những người được điều trị bằng NPH và insulin thông thường (41%) (55).
Biến chứng
Hạ đường huyết và hạ kali máu là hai biến chứng thường gặp khi điều trị quá liều DKA với insulin và bicarbonate, nhưng những biến chứng này ít xảy ra hơn khi điều trị insulin liều thấp (4,56,57). Theo dõi đường huyết thường xuyên (cứ 1-2 giờ một lần) là bắt buộc để nhận biết hạ đường huyết vì nhiều bệnh nhân DKA bị hạ đường huyết trong quá trình điều trị không có biểu hiện tăng tiết mồ hôi, hồi hộp, mệt mỏi, đói và nhịp tim nhanh. Nhiễm toan không anion gap tăng clo máu, được thấy trong giai đoạn phục hồi của DKA, tự giới hạn với một số hậu quả lâm sàng (43). Điều này có thể do mất ketoanions , được chuyển hóa thành bicarbonate trong quá trình tiến triển DKA và truyền dịch dư thừa chứa clorua trong quá trình điều trị (4).
Phù não, xảy ra ở 0,3–1,0% các đợt DKA ở trẻ em, cực kỳ hiếm ở bệnh nhân người lớn trong quá trình điều trị DKA. Phù não phổ biến với tỷ lệ tử vong 20 – 40% (5) và chiếm 57 – 87% tổng số ca tử vong do DKA ở trẻ em (70,71). Các triệu chứng và dấu hiệu của phù não có thể thay đổi và bao gồm khởi đầu đau đầu, suy giảm dần mức độ ý thức, co giật, không kiểm soát được cơ vòng, thay đổi đồng tử, phù gai thị, nhịp tim chậm, tăng huyết áp và ngừng hô hấp (71). Một số cơ chế đã được đề xuất, bao gồm vai trò của thiếu máu cục bộ / thiếu oxy não, tạo ra các chất trung gian gây viêm khác nhau (72), tăng lưu lượng máu não, gián đoạn vận chuyển ion màng tế bào, và sự thay đổi nhanh chóng dịch trong ngoại bào và nội bào dẫn đến thay đổi áp lực thẩm thấu. Phòng ngừa có thể bao gồm tránh bù dịch quá mức và giảm nhanh áp lực thẩm thấu huyết tương, giảm dần lượng đường huyết và duy trì đường huyết trong khoảng 250 – 300 mg / dl cho đến khi áp lực thẩm thấu huyết thanh của bệnh nhân được bình thường hóa và tình trạng tri giác được cải thiện. Truyền Manitol và thở máy được đề nghị để điều trị phù não (73).
Dự phòng
Nhiều trường hợp DKA và HHS có thể được ngăn ngừa bằng cách tiếp cận tốt hơn với dịch vụ chăm sóc y tế, giáo dục bệnh nhân thích hợp và giao tiếp hiệu quả với nhà cung cấp dịch vụ chăm sóc sức khỏe trong thời gian bị bệnh. Điều quan trọng nhất trong nỗ lực này là cải thiện giáo dục về quản lý “ sick day “ bao gồm những điều sau đây:
1) Liên hệ sớm với nhà cung cấp dịch vụ chăm sóc sức khỏe.
2) Nhấn mạnh tầm quan trọng của insulin trong thời gian bị bệnh và những lý do không bao giờ ngừng sử dụng mà không liên hệ với nhóm chăm sóc sức khỏe.
3) Xem xét các mục tiêu về đường huyết và việc sử dụng bổ sung insulin tác dụng ngắn hoặc tác dụng nhanh.
4) Có sẵn thuốc để hạ sốt và điều trị nhiễm trùng.
5) Bắt đầu chế độ ăn lỏng dễ tiêu hóa có chứa carbohydrate và muối khi buồn nôn.
6) Giáo dục các thành viên trong gia đình về quản lý ngày ốm và ghi chép sổ sách bao gồm đánh giá và ghi lại nhiệt độ, đường huyết, và xét nghiệm xeton trong nước tiểu / máu; insulin; nhập; và cân nặng. Tương tự, việc giám sát và giáo dục nhân viên đầy đủ trong các cơ sở dài hạn có thể ngăn chặn nhiều trường hợp nhập viện HHS do mất nước ở những người cao tuổi không thể nhận biết hoặc điều trị tình trạng đang tiến triển này.
Việc sử dụng máy đo glucose-xeton tại nhà có thể cho phép nhận biết sớm tình trạng nhiễm toan ceton sắp xảy ra, điều này có thể giúp hướng dẫn liệu pháp insulin tại nhà và có thể ngăn ngừa việc nhập viện do DKA. Ngoài ra, theo dõi xeton trong máu tại nhà, đo nồng độ beta-hydroxybutyrate trên mẫu máu lấy ngón tay, hiện đã được bán trên thị trường (37).
Nhận xét rằng việc ngừng sử dụng insulin vì lý do kinh tế là nguyên nhân phổ biến của DKA (74,75) nhấn mạnh sự cần thiết của hệ thống cung cấp dịch vụ chăm sóc sức khỏe của chúng ta để giải quyết vấn đề này, vốn tốn kém và nghiêm trọng về mặt lâm sàng. Tỷ lệ ngừng sử dụng insulin và tiền sử kém tuân thủ điều trị chiếm hơn một nửa số trường hợp nhập viện DKA ở các nhóm dân cư nội thành và thiểu số (9,74,75). Một số rào cản về văn hóa và kinh tế xã hội, chẳng hạn như tỷ lệ biết chữ thấp, nguồn tài chính hạn chế và khả năng tiếp cận chăm sóc sức khỏe hạn chế, ở những bệnh nhân nghèo trung bình có thể giải thích cho việc thiếu tuân thủ và tại sao DKA tiếp tục xảy ra với tỷ lệ cao như vậy ở bệnh nhân nội thành . Những phát hiện này cho thấy rằng phương thức cung cấp giáo dục bệnh nhân và chăm sóc sức khỏe hiện nay có những hạn chế đáng kể. Giải quyết các vấn đề sức khỏe ở người Mỹ gốc Phi và các cộng đồng thiểu số khác đòi hỏi sự công nhận rõ ràng về thực tế rằng những dân số này có thể khá đa dạng trong các phản ứng hành vi của họ đối với bệnh tiểu đường (76)
Nguồn lực đáng kể được chi cho chi phí nằm viện. Các đợt DKA đại diện cho >1 trên mỗi 4 USD được chi cho chăm sóc y tế trực tiếp cho bệnh nhân người lớn mắc bệnh tiểu đường loại 1 và cứ 2 USD ở những bệnh nhân trải qua nhiều đợt (77). Dựa trên trung bình hàng năm có 135.000 ca nhập viện vì DKA ở Hoa Kỳ, với chi phí trung bình là 17.500 USD cho mỗi bệnh nhân, chi phí bệnh viện hàng năm cho bệnh nhân DKA có thể vượt quá 2,4 tỷ USD mỗi năm (3). Một nghiên cứu gần đây (2) báo cáo rằng gánh nặng chi phí do những lần nhập viện có thể tránh được do bệnh tiểu đường không kiểm soát được trong thời gian ngắn bao gồm DKA là đáng kể (2,8 tỷ USD). Tuy nhiên, tác động lâu dài của bệnh tiểu đường không được kiểm soát và gánh nặng kinh tế của nó có thể đáng kể hơn vì nó có thể góp phần vào các biến chứng khác nhau. Bởi vì hầu hết các trường hợp xảy ra ở những bệnh nhân đã biết mắc bệnh tiểu đường và với DKA trước đó, các nguồn lực cần được chuyển hướng sang hướng phòng ngừa bằng cách tài trợ cho việc tiếp cận tốt hơn với các chương trình chăm sóc và giáo dục phù hợp với nhu cầu cá nhân, bao gồm cả niềm tin chăm sóc sức khỏe dân tộc và cá nhân. Ngoài ra, các nguồn lực cần được hướng đến việc giáo dục các nhà cung cấp dịch vụ chăm sóc ban đầu và nhân viên trường học để họ có thể xác định các dấu hiệu và triệu chứng của bệnh tiểu đường không kiểm soát được và để bệnh tiểu đường mới khởi phát có thể được chẩn đoán sớm hơn. Các nghiên cứu gần đây cho thấy rằng bất kỳ hình thức giáo dục nào về dinh dưỡng đều giúp giảm tỷ lệ nhập viện (78). Trên thực tế, các hướng dẫn về giáo dục tự quản lý bệnh tiểu đường đã được phát triển bởi một lực lượng đặc nhiệm gần đây để xác định mười tiêu chuẩn chi tiết về giáo dục tự quản lý bệnh tiểu đường (79).
Lời cảm ơn – Không có xung đột lợi ích tiềm ẩn nào liên quan đến bài viết này đã được báo cáo.
Tài liệu tham khảo
- National Center for Health Statistics. National hospital discharge and ambu- latory surgery data [article online]. Available from http://www.cdc.gov/nchs/ about/major/hdasd/nhds.htm. Accessed 24 January 2009
- KimS.Burdenofhospitalizationsprimar- ily due to uncontrolled diabetes: implica- tions of inadequate primary health care in the United States. Diabetes Care 2007;30: 1281–1282
- Agency for Healthcare Research and Quality. Databases and related tools from the healthcare cost and utilization project (HCUP) [article online]. National Center for Health Statistics, Centers for Disease Control. Available from www.hcup-us. ahrq.gov/reports/statbriefs. Accessed 24 January 2009
- KitabchiAE,UmpierrezGE,MurphyMB, Barrett EJ, Kreisberg RA, Malone JI, Wall BM. Management of hyperglycemic crisesin patients with diabetes. Diabetes Care 2001;24:131–153
- Wolfsdorf J, Glaser N, Sperling MA. Dia- betic ketoacidosis in infants, children, and adolescents: a consensus statement from the American Diabetes Association. Diabetes Care 2006;29:1150 –2259
- White NH. Diabetic ketoacidosis in chil- dren. Endocrinol Metab Clin North Am 2000;29:657– 682
- Graves EJ, Gillium BS, the National Cen- ter for Health Statistics. Detailed diag- noses and procedures: National Hospital Discharge Survey, 1995. Vital Health Stat 13 1997;(130):1–146
- Malone ML, Gennis V, Goodwin JS. Char- acteristics of diabetic ketoacidosis in older versus younger adults. J Am Geriatr Soc 1992;40:1100 –1104
- Umpierrez GE, Kelly JP, Navarrete JE, Casals MM, Kitabchi AE. Hyperglycemic crises in urban blacks. Arch Intern Med 1997;157:669 – 675
- Ennis ED, Stahl EJVB, Kreisberg RA. The hyperosmolar hyperglycemic syndrome. Diabetes Rev 1994;2:115–126
- Lorber D. Nonketotic hypertonicity in di- abetes mellitus. Med Clin North Am 1995;79:39 –52
- Kitabchi AE, Fisher JN, Murphy MB, Rumbak MJ. Diabetic ketoacidosis and the hyperglycemic hyperosmolar nonke- totic state. In Joslin’s Diabetes Mellitus. 13th ed. Kahn CR, Weir GC, Eds. Phila- delphia, Lea & Febiger, 1994, p. 738– 770
- Kitabchi AE, Umpierrez GE, Murphy MB, Kreisberg RA. Hyperglycemic crises in adult patients with diabetes. Diabetes Care 2006;29:2739 –2748
- DeFronzo RA, Matzuda M, Barret E. Dia- betic ketoacidosis: a combined metabolic- nephrologic approach to therapy. Diabetes Rev 1994;2:209 –238
- Luzi L, Barrett EJ, Groop LC, Ferrannini E, DeFronzo RA. Metabolic effects of low- dose insulin therapy on glucose metabo- lism in diabetic ketoacidosis. Diabetes 1988;37:1470 –1477
- van de Werve G, Jeanrenaud B. Liver gly- cogen metabolism: an overview. Diabetes Metab Rev 1987;3:47–78
- Felig P, Sherwin RS, Soman V, Wahren J, Hendler R, Sacca L, Eigler N, Goldberg D, Walesky M. Hormonal interactions in the regulation of blood glucose. Recent Prog Horm Res 1979;35:501–532
- Barrett EJ, DeFronzo RA, Bevilacqua S, Ferrammi E. Insulin resistance in diabetic ketoacidosis. Diabetes 1982;31:923–928
- Miles JM, Haymond MW, Nissen S, Gerich JE. Effects of free fatty acid avail- ability, glucagon excess and insulin defi- ciency on ketone body production in postabsorptive man. J Clin Invest 1983; 71:1554 –1561
- 20. Stentz FB, Umpierrez GE, Cuervo R, Kitabchi AE. Proinflammatory cytokines,
- 33.markers of cardiovascular risks, oxidative stress, and lipid peroxidation in patients with hyperglycemic crises. Diabetes 2004; 53:2079 –2086
- Buyukasik Y, Ileri NS, Haznedaroglu IC, Karaahmetoglu S, Muftuoglu O, Kirazli S, Dundar S. Enhanced subclinical coagula- tion activation during diabetic ketoacido- sis. Diabetes Care 1998;21:868 – 870 Delaney MF, Zisman A, Kettyle WM. Di- abetic ketoacidosis and hyperglycemic hyperosmolar nonketotic syndrome. En- docrinol Metab Clin North Am 2000;29: 683–705 Hormones 1972;3:36 – 41
- Umpierrez G, Freire AX. Abdominal pain in patients with hyperglycemic crises. J Crit Care 2002;17:63– 67
- Campbell IW, Duncan LJ, Innes JA, Mac- Cuish AC, Munro JF. Abdominal pain in diabetic metabolic decompensation: clin- ical significance. JAMA 1975;233:166 –168
- Kitabchi AE, Fisher JN. Insulin therapy of diabetic ketoacidosis: physiologic versus pharmacologic doses of insulin and their routes of administration. In Handbook of Diabetes Mellitus. Brownlee M, Ed. New York, Garland ATPM Press, 1981, p. 95– Peden NR, Broatan JT, McKenry JB. Dia- 149 betic ketoacidosis during long-term treat- ment with continuous subcutaneous insulin infusion. Diabetes Care 1984;7:1–5 Weissberg-Benchell J, Antisdel-Lomaglio J, Seshadri R. Insulin pump therapy: a meta-analysis. Diabetes Care 2003;26: 1079 –1087 Wachtel TJ, Silliman RA, Laberton P. Pre- disposing factor for the diabetic hyperos- molar state. Arch Intern Med 1987;147: 499 –501 Avella J, Wetli CV, Wilson JC, Katz M, Hahn T. Fatal olanzapine-induced hyer- glycemic ketoacidosis. Am J Forensic Med Pathol 2004;25:172–175
- Sheikh-Ali M, Karon BS, Basu A, Kudva YC, Muller LA, Xu J, Schwenk WF, Miles JM. Can serum ?-hydroxybutyrate be used to diagnose diabetic ketoacidosis? Diabetes Care 2008;31:643– 647
- Miles JM, Gerich JE. Glucose and ketone body kinetics in diabetic ketoacidosis. Clin Endocrinol Metab 1983;12:303–319
- Munro JF, Campbell IW, McCuish AC, Duncan LJ. Euglycaemic diabetic ketoac- idosis. Br Med J 1973;2:578 –580
- Burge MR, Hardy KJ, Schade DS. Short- term fasting is a mechanism for the de- velopment of euglycemic ketoacidosis during periods of insulin deficiency. J Clin Endocrinol Metab 1993;76:1192– Campanella LM, Lartey R, Shih R. Sever hyperglycemic hyperosmolar nonketotic 1198 coma in a nondiabetic patient receiving aripiprazole. Am Emerg Med 2009;53: 264 –266 Umpierrez GE, Smiley D, Kitabchi AE. Narrative review: ketosis-prone type 2 di- abetes mellitus. Ann Intern Med 2006; 144:350 –357 Umpierrez GE, Woo W, Hagopian WA, Isaacs SD, Palmer JP, Gaur LK, Nepom GT, Clark WS, Mixon PS, Kitabchi AE. Immunogenetic analysis suggest different pathogenesis between obese and lean Af- rican-Americans with diabetic ketoacido- sis. Diabetes Care 1999;22:1517–1523 Maldonado M, Hampe CS, Gaur LK, D’Amico S, Iyer D, Hammerle LP, Bolgiano D, Rodriguez L, Rajan A, Lernmark A, Bala- subramanyam A. Ketosis-prone diabetes: dissection of a heterogeneous syndrome us- ing an immunogenetic and beta-cell func- tional classification, prospective analysis, and clinical outcomes. J Clin Endocrinol Metab 2003;88:5090 –5098 Mauvais-Jarvis F, Sobngwi E, Porcher R, Riveline JP, Kevorkian JP, Vaisse C, Char- pentier G, Guillausseau PJ, Vexiau P, Gautier JF. Ketosis-prone type 2 diabetes in patients of sub-Saharan African origin: clinical pathophysiology and natural his- tory of ?-cell dysfunction and insulin re- sistance. Diabetes 2004;53:645– 653 Balasubramanyam A, Nalini R, Hampe CS, Maldonado M. Syndromes of ketosis- prone diabetes mellitus. Endocr Rev 2008; 29:292–302
- Matz R. Hypothermia in diabetic acidosis.
- Slovis CM, Mork VG, Slovis RJ, Bain RP. Diabetic ketoacidosis and infection: leu- kocyte count and differential as early pre- dictors of serious infection. Am J Emerg Med 1987;5:1–5
- Razavi L, Kitabchi AE, Stentz FB, Wan JY, Larijani BA, Tehrani M, Gozashti M, Mid- far K, Taheri E. Proinflammatory cyto- kines in response to insulin-induced hypoglycemic stress in healthy subjects. Metab Clin Exp 2009;58:443– 448
- Adrogue HJ, Wilson H, Boyd AE 3rd, Suki WN, Eknoyan G. Plasma acid-base pat- terns in diabetic ketoacidosis. N Engl J Med 1982;307:1603–1610
- Rumbak MJ, Hughes TA, Kitabchi AE. Pseudonormoglycemia in diabetic keto- acidosis with elevated triglycerides. Am J Emerg Med 1991;9:61– 63
- Kaminska ES, Pourmotabbed G. Spurious laboratory values in diabetic ketoacidosis and hyperlipidemia. Am J Emerg Med 1993;11:77– 80
- Fisher JN, Kitabchi AE. A randomized study of phosphate therapy in the treat- ment of diabetic ketoacidosis. J Clin En- docrinol Metab 1983;57:177–180
- Fisher JN, Shahshahani MN, Kitabchi AE. Diabetic ketoacidosis: low-dose insulin therapy by various routes. N Engl J Med 1977;297:238 –241
- Yadav D, Nair S, Norkus EP, Pitchumoni CS. Nonspecific hyperamylasemia and hyperlipasemia in diabetic ketoacidosis: incidence and correlation with biochem- ical abnormalities. Am J Gastroenterol2000;95:3123–3128
- Umpierrez GE, DiGirolamo M, Tuvlin JA,
- Isaacs SD, Bhoola SM, Kokko JP. Differ- ences in metabolic and hormonal milieu in diabetic- and alcohol-induced ketoaci- dosis. J Crit Care 2000;15:52–59
- Nyenwe E, Loganathan R, Blu ̈ m S, Ezuteh D, Erani D, Wan J, Palace MR, Kitabchi AE. Active use of cocaine: an independent risk factor for recurrent diabetic ketoaci- dosis in a city hospital. Endocr Pract 2007;13:22–29
- Soveid M, Ranjbar-Omrani G. Ketoacido- sis as the primary manifestation of acro-
- megaly. Arch Iranian Med 2005;8:326 – 328
- Kuzuya T, Matsuda A, Sakemoto Y, Yamamoto K, Saito T, Yoshida S. A case of pituitary gigantism who had two episodes of diabetic ketoacidosis followed by com- plete recovery of diabetes. Endocrinol Jpn 1983;30:323–334
- Hillman K. Fluid resuscitation in diabetic emergencies: a reappraisal. Intensive Care Med 1987;13:4 – 8
- Bratusch-Marrain PR, Komajati M, Waldhausal W. The effect of hyperos- molarity on glucose metabolism. Pract Cardiol 1985;11:153–163
- Umpierrez GE, Jones S, Smiley D, Mulli- gan P, Keyler T, Temponi A, Semakula C, Umpierrez D, Peng L, Cero ́ n M, Robalino G. Insulin analogs versus human insulin in the treatment of patients with diabetic ketoacidosis: a randomized controlled trial. Diabetes Care 2009;32:1164 –1169
- Alberti KGGM, Hockaday TDR, Turner RC. Small doses of intramuscular insulin in the treatment of diabetic ‘coma.’ Lancet 1973;5:515–522
- Kitabchi AE, Ayyagari V, Guerra SNO, Medical House Staff. Efficacy of low dose vs conventional therapy of insulin for treatment of diabetic ketoacidosis. Ann In- tern Med 1976;84:633– 638
- Kitabchi AE, Umpierrez GE, Fisher JN, Murphy MB, Stentz FB. Thirty years of personal experience in hyperglycemic cri- ses: diabetic ketoacidosis and hyperglyce- mic hyperosmolar state. J Clin Endocrinol Metab 2008;93:1541–1552
- Kitabchi AE, Murphy MB, Spencer J, Mat- teri R, Karas J. Is a priming dose of insulin
- necessary in a low-dose insulin protocol for the treatment of diabetic ketoacidosis? Diabetes Care 2008;31:2081–2085
- Umpierrez GE, Latif K, Stoever J, Cuervo R, Park L, Freire AX, Kitabchi AE. Efficacy of subcutaneous insulin lispro versus con- tinuous intravenous regular insulin for the treatment of diabetic ketoacidosis. Am J Med 2004;117:291–296
- Umpierrez GE, Latif KA, Cuervo R, Kara- bell A, Freire AX, Kitabchi AE. Treatment of diabetic ketoacidosis with subcutane- ous insulin aspart. Diabetes Care 2004; 27:1873–1878
- Viallon A, Zeni F, Lafond P, Venet C, Tardy B, Page Y, Bertrand JC. Does bicar- bonate therapy improve the management of severe diabetic ketoacidosis? Crit Care Med 1999;27:2690 –2693
- Mitchell JH, Wildenthal K, Johnson RL Jr. The effects of acid-base disturbances on cardiovascular and pulmonary function. Kidney Int 1972;1:375–389
- Morris LR, Murphy MB, Kitabchi AE. Bi- carbonate therapy in severe diabetic keto- acidosis. Ann Intern Med 1986;105:836 – 840
- Glaser N, Barnett P, McCaslin I, Nelson D, Trainor J, Louie J, Kaufman F, Quayle K, Roback M, Malley R, Kuppermann N, the Pediatric Emergency Medicine Collabora- tive Research Committee of the American Academy of Pediatrics. Risk factors for ce- rebral edema in children with diabetic ke- toacidosis. N Engl J Med 2001;344:264 – 269
- Latif KA, Freire AX, Kitabchi AE, Umpi- errez GE, Qureshi N. The use of alkali therapy in severe diabetic ketoacidosis. Diabetes Care 2002;25:2113–2114
- Winter RJ, Harris CJ, Phillips LS, Green OC. Diabetic ketoacidosis: induction of hypocalcemia and hypomagnesemia by phosphate therapy. Am J Med 1979;67: 897–900
- Kreisberg RA. Phosphorus deficiency and hypophosphatemia. Hosp Pract 1977;12: 121–128
- Miller DW, Slovis CM. Hypophos- phatemia in the emergency department therapeutics. Am J Emerg Med 2000;18: 457– 461
- Rosenbloom AL. Intracerebral crises dur- ing treatment of diabetic ketoacidosis. Diabetes Care 1990;13:22–33 Marcin JP, Glaser N, Barnett P, McCaslin
- I, Nelson D, Trainor J, Louie J, Kaufman F, Quayle K, Roback M, Malley R, Kup- permann N. Factors associated with ad- verse outcomes in children with diabetic ketoacidosis-related cerebral edema. J Pe- diatr 2002;141:793–797
- Abbott NJ. Inflammatory mediators and modulation of blood-brain barrier perme- ability. Cell Mol Neurobiol 2000;20:131– 147
- Roberts MD, Slover RH, Chase HP. Dia- betic ketoacidosis with intracerebral com- plications. Pediatr Diabetes 2001;2:109 – 114
- Musey VC, Lee JK, Crawford R, Klatka MA, McAdams D, Phillips LS. Diabetes in urban African-Americans. I. Cessation of insulin therapy is the major precipitating cause of diabetic ketoacidosis. Diabetes Care 1995;18:483– 489
- Maldonado MR, Chong ER, Oehl MA, Balasubramanyam A. Economic impact of diabetic ketoacidosis in a multiethnic in- digent population: analysis of costs based on the precipitating cause. Diabetes Care 2003;26:1265–1269
- Anderson RM, Herman WH, Davis JM, Freedman RP, Funnell MM, Neighbors HW. Barriers to improving diabetes care for blacks. Diabetes Care 1991;14:605– 609
- Javor KA, Kotsanos JG, McDonald RC, Baron AD, Kesterson JG, Tierney WM. Di- abetic ketoacidosis charges relative to medical charges of adult patients with type I diabetes. Diabetes Care 1997;20: 349 –354
- RobbinsJM,ThatcherGE,WebbDA,Val- manis VG. Nutritionist visits, diabetes classes, and hospitalization rates and charges: the Urban Diabetes Study. Dia- betes Care 2008;31:655– 660
- Funnell MM, Brown TL, Childs BP, Haas LB, Hosey GM, Jensen B, Maryniuk M, Peyrot M, Piette JD, Reader D, Siminerio LM, Weinger K, Weiss MS. National Stan- dards for Diabetes Self-Management Edu- cation. Diabetes Care 2009;32(Suppl. 1): S87–S94
Để lại một phản hồi